EULAR recommendations for the management of AAV - EGPA -
11.organ-threatening or life-threatening な症状を呈する、新規or再発性EGPAの寛解導入療法において、高用量GCとCYCとの併用療法を推奨する。代替療法として高用量GCとRituximabの併用も考慮されてよい。
12.organ-threatening or life-threatening な症状を呈さない、新規or再発性のEGPAの寛解導入療法においてGCによる治療を推奨する。
13.organ-threatening or life-threatening な症状を呈さない、再発性or抵抗性のEGPAの寛解導入療法において、Mepolizumabの使用を推奨する。
14.organ-threatening or life-threatening な症状を呈する、EGPAの維持療法として、MTX, AZA, Mepolizumab, Rituximabを考慮すべき。
↓解説文
For induction of remission in new-onset or relapsing EGPA with organ-threatening or life-threatening manifestations, we recommend treatment with a combination of high-dose GCs and CYC. A combination of high-dose GCs and RTX may be considered as an alternative.
Since the last update of these recommendations, results of three RCTs enrolling only or mainly patients with EGPA have been reported,77 78 173 which allowed the development of separate recommendations for EGPA for this update (figure 3). The Five Factor Score (FFS) is used for prognostic assessment of EGPA.174 Particularly, cardiac involvement has been associated with increased mortality in EGPA.36 175 176 However, under optimised management in centres of expertise, the prognosis of cardiac involvement appears to be better than previously reported. This may reflect more frequent diagnosis of milder forms of cardiac disease through use of cardiac MRI and greater awareness among physicians regarding cardiac disease in EGPA.177 In a recent series from the French Vasculitis Study Group of 70 patients with EGPA with cardiac manifestations treated with high-dose GC, mostly along with CYC, no patient died as a consequence of cardiac involvement during a 10-year observation period.178 With the aim of preventing permanent organ damage due to EGPA, patients with severe involvement of the kidneys, central and peripheral nervous system, or gastrointestinal tract are also considered to be candidates for treatment with CYC (table 2).179 In a randomised, open-label trial in patients with EGPA and poor prognosis (FFS ≥1), 12 compared with 6 pulsed doses of CYC were associated with a lower rate of minor relapses but did not improve response rate or reduce severe relapses.180 Therefore, we recommend treatment be switched to a less intensive remission maintenance therapy after six pulses of CYC if remission is achieved, and the GC dose is reduced by then to approximately 7.5 mg per day (see online supplemental table 2 for protocols).
An RCT examining the use of RTX in EGPA (REOVAS) included 105 patients with new-onset or relapsing EGPA of whom 42 had life-threatening or organ-threatening disease (FFS ≥1) (abstract).77 Patients with FFS ≥1 received high-dose GCs plus either 2×1 g RTX (days 1 and 15) or nine pulses of CYC over 13 weeks. The primary endpoint of on-treatment remission was reached at similar frequencies at days 180 and 360 in both groups, but the limited number of patients, the superiority design and the lack of fully published results do not allow for strong conclusions regarding non-inferiority. Adverse events, cumulative prednisone doses and quality of life were not different between groups. Results were similar in both newly diagnosed and relapsing disease. In contrast to earlier observational studies,181 182 the response to RTX was not higher in MPO-ANCA- positive patients compared with ANCA-negative patients, consistent with consensus recommendations on ANCA testing that treatment decisions in EGPA should not be influenced solely by ANCA status.76 Keeping in mind that the results of the REOVAS trial have not been fully published yet, the data reported so far are deemed sufficiently strong to consider RTX as an alternative to CYC, particularly in patients in which exposure to CYC needs to be avoided, and are consistent with earlier observational reports (see recommendation no. 3). In contrast to GPA and MPA, no studies have compared different GC tapering strategies in the treatment of EGPA. In the absence of data to support an evidence-based recommendation on GC tapering in EGPA, recommendations made for GPA and MPA (statement no. 4) can be used as an orientation. However, asthma and ear, nose and throat (ENT) exacerbation increase the GC requirement in patients with EGPA, leading to prolonged tapering.183 Therefore, interdisciplinary management involving pulmonologists and/or otorhinolaryngologists aimed at optimising treatment (including topical agents) of asthma, polyposis and sinusitis is recommended.
EGPAにおけるRTXの使用を検討したRCT(REOVAS)には、新規発症または再発のEGPA患者105名が含まれ、そのうち42名が生命を脅かすか臓器を脅かす疾患(FFS≧1)でした(abstrac)77。FFS≧1の患者には、高用量のGCと2×1g RTX(1日と15日)または13週間のCYC9パルスのいずれかを投与した。主要評価項目である治療中寛解は、両群とも180日目と360日目に同程度の頻度で到達したが、患者数が限られていること、優越性デザインであること、完全に出版されていないことから、非劣性に関する強い結論は得られない。有害事象、PSNの累積投与量、QOLに群間差はなかった。結果は、新規に診断された疾患と再発した疾患の両方で同様であった。以前の観察研究とは対照的に、181 182 MPO-ANCA 陽性患者では、ANCA 陰性患者と比較して RTX に対する反応性は高くなく、EGPA における治療決定は ANCA の状態のみによって影響されるべきではないという ANCA 検査に関する合意勧告と一致している76。REOVAS試験の結果がまだ完全に発表されていないことを考慮すると、これまでに報告されたデータは、特にCYCへの曝露を避ける必要がある患者において、RTXをCYCの代替薬として検討するのに十分な強度を持つと考えられ、以前の観察報告と一致している(recommendation no. 3)。GPAやMPAとは対照的に、EGPAの治療において異なるGC漸減戦略を比較した研究はない。EGPAにおけるGC漸減についてエビデンスに基づく推奨を支持するデータがない場合、GPAおよびMPAについてなされた推奨(声明No.4)を参考とすることができる。しかし、喘息や耳鼻咽喉科の増悪は、EGPA患者におけるGC要求必要量を増加させ、漸減の長期化につながる。
For induction of remission in new-onset or relapsing EGPA without organ-threatening or life-threatening manifestations, we recommend treatment with GCs.
Patients with EGPA without adverse prognostic factors (FFS=0) treated with GC only achieve remission >90% of the time, but relapses are common once GCs are tapered.174 184 Therefore, clinicians frequently combine GCs with other immunosuppressants or biologics. However, the SLR revealed that evidence supporting GC-sparing therapy in newly diagnosed patients with EGPA without organ-threatening or life-threatening manifestations is low.10 A prospective placebo-controlled study showed that therapy with AZA for 1 year in addition to GC had no effect on the risk of relapse, cumulative GC requirement, or the rate of asthma and sinusitis exacerbation compared with GC monotherapy in EGPA without poor prognostic factors (FFS=0).173 Recent long-term study data also showed that, within 5 years, 48% of all patients experienced vasculitis relapses, and prior therapy with AZA did not reduce this risk.184 The REOVAS trial included 63 new-onset or relapsing patients with EGPA with an FFS of 0 who were randomised to receive high-dose GCs together with either 2×1 g RTX (days 1 and 15) or placebo. Efficacy and safety outcomes after 180 and 360 days were not different between RTX and placebo group. Because the trial was not designed as a non-inferiority trial and since it has been reported only in abstract format so far, the data preclude from making strong conclusions but provide no support for use of RTX for remission induction in this subgroup of patients. No RCTs are available on the use of MTX, MMF or leflunomide in EGPA. Small observational studies on the use of MTX or MMF did not include control groups and carry a high risk of bias.185 186 As the evidence to support immunosuppression beyond GCs in new-onset EGPA without risk factors for worse outcome is low,10 decisions on the use of GC-sparing therapy in this subset of patients may be made on an individual basis considering risk factors of GC-related morbidity.
For induction of remission in patients with relapsing or refractory EGPA without active organ-threatening or life-threatening disease, we recommend the use of mepolizumab.
The IL-5 inhibitor mepolizumab was evaluated in a randomised double-blind placebo-controlled phase III study (MIRRA) that included 136 patients with relapsing or refractory EGPA with disease duration of at least 6 months.78 The study protocol allowed the inclusion of patients without vasculitic manifestations, while patients with active life-threatening or organ-threatening manifestations were excluded. After treatment with prednisolone at a stable dose ≥7.5 mg per day prior to baseline, 54% of patients in the mepolizumab arm and 71% of patients in the control arm had active disease at randomisation with a Birmingham Vasculitis Activity Score (BVAS) >0. Patients were randomised 1:1 to continuation of standard therapy (including other conventional immunosuppressive agents in more than 50% of patients in each arm) or standard therapy plus mepolizumab at a dose of 300 mg subcutaneously every 4 weeks. Both co-primary endpoints (the number of weeks in remission on a prednisolone dose reduced to 4 mg and the proportion of patients in remission at weeks 36 and 48) were met in favour of mepolizumab.78 A post-hoc analysis showed that treatment with mepolizumab was associated with additional clinically relevant endpoints, such as remission and GC reduction of >50%, in over half of the patients treated with mepolizumab.187 Based on its association with clinically meaningful improvement of disease control and reduction of GC demand, and good safety profile compared with conventional immunosuppressants, this task force recommends the use of mepolizumab with relapsing or refractory, non-organ- threatening or life-threatening EGPA. Mepolizumab has recently been approved for this indication in many European countries. Data from studies using mepolizumab for treatment of life- threatening or organ-threatening or new-onset EGPA are currently lacking.10
Other IL-5 or IL-5 receptor inhibitors (reslizumab, benralizumab) showed efficacy in small open-label pilot studies in EGPA,188 189 but data from RCTs using these agents are not yet available. In a retrospective multicentre series, anti-IgE- targeted therapy with omalizumab appeared to be less effective than mepolizumab.190 As discussed above, data showing improved outcomes with other biological or conventional drugs for non-severe relapsing or refractory EGPA are lacking. In patients for whom mepolizumab is not effective or not tolerated, AZA, MTX, MMF or RTX can be considered on an individual basis.184–186 190–192
A true refractory course of EGPA with life-threatening and organ-threatening manifestations is rare if patients are treated with high-dose GC and a CYC-based or RTX-based induction regimen.77 180 Data guiding treatment decisions in this small subgroup of patients are scarce, and true refractory severe EGPA needs to be carefully distinguished from infections and comorbidities. Therefore, patients with suspected refractory severe EGPA should be managed at centres of expertise.
For maintenance of remission of relapsing EGPA after induction of remission for non-organ- threatening or life-threatening manifestations at the time of relapse, we recommend treatment with mepolizumab. For maintenance of remission of EGPA after induction of remission for organ-threatening or life-threatening disease, treatment with MTX, AZA, mepolizumab or RTX should be considered.
In view of the relapsing nature of EGPA requiring long-term use of GCs in most patients, other agents for maintenance of remission are commonly prescribed in an attempt to be GC sparing. In an RCT (MIRRA, see recommendation no. 13 for details) that enrolled patients with EGPA who had relapsing or refractory disease, rates of severe and non-severe relapses were significantly lower and the median GC dose throughout the study was lower in the mepolizumab group compared with the control group.78 Adverse events occurred at similar rates in the mepolizumab group and the placebo group, while serious adverse events were somewhat more common in the placebo group (18% vs 26%). In view of its efficacy and good safety profile, the use of mepolizumab after induction of remission for non-organ- threatening or life-threatening manifestations at the time of relapse is recommended. In an RCT that enrolled 51 patients with EGPA and no organ-threatening or life-threatening manifestations, AZA for 1 year in addition to GC had no effect on the risk of relapse, cumulative GC requirement, or the rate of asthma and sinusitis exacerbation compared with GC monotherapy. 173 There is little evidence to recommend routine use of other immunomodulatory agents for maintenance of remission in EGPA without organ-threatening or life-threatening manifestations.10
The SLR identified only one prospective study addressing remission maintenance strategies in patients with EGPA who attained remission after treatment for life-threatening or organ-threatening disease.10 A single-centre prospective randomised trial compared oral CYC with MTX for 1 year after remission induction with CYC in different subtypes of AAV.193 In the subgroup of 30 patients with EGPA who had either an FFS >1 or peripheral neuropathy, no difference in relapse rates between the two treatment arms was observed. Although no excess in adverse events was found in this study, we do not recommend CYC for remission maintenance in view of its toxicity. However, this study provides a rationale for use of MTX for maintenance of remission in EGPA, although the small sample size of patients with EGPA precludes a strong recommendation. As observational studies reported favourable outcomes on the use of AZA, mepolizumab and RTX for maintenance of remission,36 190 these agents can also be considered for remission maintenance in EGPA after induction of remission for organ-threatening or life-threatening manifestations. In view of its efficacy in eosinophilic asthma, mepolizumab should also be considered for patients with EGPA with residual GC-dependent asthma who achieved remission of major organ involvement.
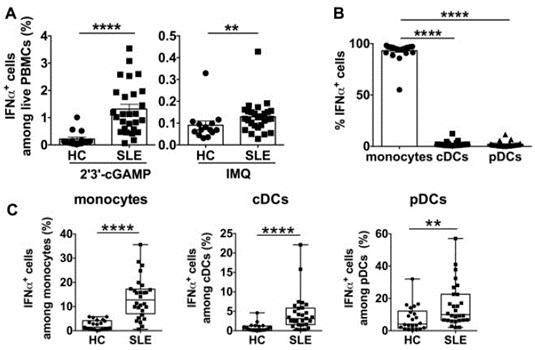
我々は、2’3’-cGAMPによるcGAS-STING経路刺激後の骨髄系細胞のIFNa産生能について調査した。ループス単球はcGAS-STING経路の活性化によりIFNaを産生し、IFNa産生単球の頻度はSLE疾患活動性と正の相関があった。さらに、STINGの発現とTBK1などの下流分子との共局在が増加した。IFNaへの先行曝露は、単球のIFNa産生能を増強した。最後に、mTOR (mechanistic target of rapamycin) 経路を阻害すると、単球のIFNa産生が抑制され、ループス単球におけるSTINGとその下流分子の発現増加がダウンレギュレートされた。
Methods
Human samples
フローサイトメトリー解析には、SLEと診断された患者36人[女性34人、男性2人、年齢 (四分位範囲) 中央値39.6歳 (22.0-50.0) ]と健康対照 (HC) 26人[女性24人、男性2人、年齢中央値38歳 (23.0-47.0) ] (Table 1) 。western blotting解析では、SLE患者4名とHC3名を対象とした (Supplementary Table S1, available at Rheumatology online) 。共焦点顕微鏡による解析は、SLE患者15名とHC11名を対象とした (Supplementary Table S2, available at Rheumatology online) 。末梢血は、順天堂大学の地方倫理委員会のガイドラインに従ってインフォームドコンセントを得た後、SLE患者およびHCから採取した。SLEは、米国リウマチ学会のSLEの基準に従って診断された。疾患活動性はSLE疾患活動性指標2000 (SLEDAI-2K) により評価した。HCsは自己免疫疾患の既往がなく、免疫抑制療法を受けたことがない者とした。ヒトを対象とした研究に関する倫理指針に従って、すべてのSLE患者およびHCからICを取得した。患者の年齢、性別、治療内容はTable 1に示す。新鮮なPBMCは、BD Vacutainer CPT Mononuclear Cell Preparation Tubes with Sodium Heparin (BD Biosciences, Franklin Lakes, NJ, USA) を用いて密度勾配遠心法により全血から分離された。
Cell culture 細胞培養
PBMCは、10%ウシ胎児血清、2mM L-グルタミン、50U/mlペニシリン、50lg/mlストレプトマイシン (すべてThermo Fisher Scientific製) を含むRPMI 1640培地 (Thermo Fisher Scientific, MA, USA) 中、96-well flat-bottom platesで、5%CO2培養器中37℃で培養した。PBMCをrecombinant human IL-3 (100ng/ml;PeproTech, Rocky Hill, NJ, USA) 、TLR7アゴニスト、imiquimod (R837) (100ng/ml;InvivoGen, San Diego, CA, USA) または2’3’-cGAMP (50 lg/ml; InvivoGen) により6時間刺激を受けた。細胞内サイトカイン染色には、GolgiPlug (100ng/ml BD Biosciences) を刺激の最後の3時間の間に添加してサイトカイン分泌をブロックした。IFNa前処理実験では、PBMCをIFNa (100U/ml) (R&D Systems) で24時間培養した後、2’3’-cGAMPで刺激した。シグナル伝達分子の細胞内染色のために、PBMCをIFNa (100U/ml) (R&D Systems) または2’3’-cGAMP (50lg/ml) で2時間培養し、PBMCまたは選別単球を2’3’-cGAMP (50mg/ml) の存在または不在下でRapamycin (100nM) (ENZO Life Sciences, Plymouth Meeting, PA, USA) で培養した。
Flow cytometry
細胞染色はZombie YellowTM Fixable Viability Kit (BioLegend, San Diego, CA, USA) を用い、ヒト細胞表面抗原に対するモノクローナル抗体のcombinationを用いた:抗CD11c-Alexa700、抗HLADR-V500、抗CD19-APC-H7 (BD Biosciences) 、抗CD14-ECD、抗CD56-APC (Beckman Coulter, Brea, CA, USA) 、抗CD123-FITC、抗CD123-PV421、抗 CD3-PerPCy5. 5、抗CD3-PV605、抗CD56-PV421、抗CD56-PerCPCy5.5 (BioLegend) 、抗CD19-PE (TONBO Biosciences, San Diego, CA, USA) 。次に以下を用いて細胞内染色を行った;BD Cytofix/Cytoperm Fixation/Permeabilization Solution Kit (BD Biosciences) and anti-STING-Alexa Fluor 647 (BD Biosciences) , anti-IFNa- APC (Miltenyi Biotec, Bergisch Gladbach, Germany) , anti-IRF3-Alexa Fluor 647 (BD Biosciences) , anti-Phospho- TBK1 (pTBK1) -PE (Cell Signalling Technology, Danvers, MA, USA) , anti-Phospho-mTOR-PE (eBioscience, San Diego, CA, USA) or isotype control antibodies.単球はCD3-CD19-CD14+として同定した。cDCはCD3-CD19-CD14-CD56-HLADR+CD11c+として、pDCはCD3-CD19-CD14-CD56-HLADR+CD11c-CD123+として同定した。データはFACS LSR Fortessa (BD Biosciences) で取得し、FlowJoソフトウェア (TreeStar Inc.、Ashland, OR, USA) を使用して解析した。
IFNa measurement
IFNa濃度は、VeriKine-HS Human IFNa Alpha All Subtype ELISA Kit (PBL Assay Science, Piscataway Township, NJ, USA) を用いて、製造者の説明書に従って測定した。
Confocal microscopy 共焦点顕微
manufacturer’s instructionsに従って、抗CD14マイクロビーズ (Miltenyi Biotec) を用いてPBMCから単球を精製した。IFNa、Rapamycinまたは2’3’-cGAMPで前処理した/しない精製単球を、Thermo Shandon Cytospin 4 (Thermo Fisher Scientific) を用いて顕微鏡スライドに紡ぎ出した。単球を4%パラホルムアルデヒドで固定し、Triton X-100 (PBS中0.2%Triton X-100) で透過化した。非特異的な背景染色は、Image-iT FX Signal Enhancer (Thermo Fisher Scientific) によってブロックされた。細胞を一次抗体:anti-STING (R&D Systems, Minneapolis, MN, USA) 、anti-TBK1 (Abcam, Cambridge, UK) およびanti-LC3B (Sigma-Aldrich, St.Louis, MO, USA) とともにインキュベートした後、洗浄して二次抗体とともにインキュベートした: Alexa488-donkey抗マウスIgG、Alexa594-ドンキー抗ヤギIgG、Alexa647-ドンキー抗ウサギIgG (Jackson ImmunoResearch Laboratories, West Grove, PA, USA) 。細胞核はDAPI (Sigma-Aldrich) で染色し、Fluoromount/ Plus (Diagnostic BioSystems, Pleasanton, CA, USA) でマウントした。サンプルはFM1000D共焦点レーザー走査型顕微鏡 (オリンパス、東京、日本) で可視化し、FV10-ASWビューア (オリンパス) で解析した。共焦点解析のために、10個の単球の平均ピアソン相関係数をImageJで計算した。
Western blot analysis
単球はanti-CD14 microbeadsを用いてPBMCから精製した。HCおよびSLE患者からの単球は、Halt Protease Inhibitor Cocktail, EDTA-Free (Thermo Fisher Scientific) を含むM-PER Reagentで溶解された。タンパク質濃度は、Coomassie Plus Protein Assayキットを用いて、manufacturer’s instructionsに従って定量した。次に、4-15% Mini-PROTEAN TGX Precast Protein Gels (Bio-Rad) にサンプルあたり3.75 mgの総蛋白質を負荷した。The lysates were subjected to SDS–PAGE, and western blotting was performed with anti-STING rabbit mAb (Cell Signalling Technology) and anti-b-actin pAb (MBL). lysetes?をSDS-PAGEに供し、anti-STING rabbit mAb (Cell Signalling Technology) およびanti-b-actin pAb (MBL) を用いてwestern blottingを実施した。ブロットは、manufacturer’s instructions (Thermo Fisher Scientific)に従い、SuperSignal West Dura Extended Duration Substrateを用いた強化されたchemiluminescence assayで可視化した 。シグナルは、LAS 4000 (GE Healthcare) を用いて検出し、ImageJを用いて定量化した。
Statistical analysis
データはGraphPad Prism (GraphPad, Inc., La Jolla, CA, USA) を用いて解析し、グループ間の差はKruskal-Wallis test followed by Dunn's multiple comparisons test、Mann-Whitney U test、一元配置分散分析と Tukey multiple comparison test、Unparent t test、またはpaired t testによって解析された。有意水準は、P <0.05。2つの変数間の関連は、スピアマンの相関によって分析された。
Results
cGAS-STING stimulation enhances IFNa from SLE monocytes, cDCs and pDCs
治療に関わらずSLEにおける経時的な疾患活動性のvariationは良く認識されている。縦断的な研究が行われ、SLEにおける3つの疾患活動性のパターンが判明した;すなわち再発寛解型relapsing remitting (RRD), 長期安定型long quiescent (LQD)、慢性活動型chronic active disease (CAD)である LQDは30~40%、CADとRRDは残りの60~70%。私たちは自身のコホートにおいてSLEの標準治療を行っていて、年に3回以上受診している外来患者を対象に疾患活動性のパターンを調査した。血清は疾患活動性を評価する際に省くこととした。その結果、純粋な臨床的活動性に焦点を当てた。その結果、以下のパターンが同定された;clinically quiescent disease (CQD; SLEDAI-2K= 0 in three annual visits); CAD (SLEDAI-2K ≥ 2 in at least two out of three annual visits); RRD (SLEDAI-2K ≥ 2 in one out of three annual visits)。4つ目のパターンとしてSLEDAI-2K = 1で定義されるminimal persistent disease activity (MDA)があった。私たちのコホートから得られた結果は過去の研究と一致しており、治療中の患者の50%が三年寛解、残り50%がいくらかの活動性を有したすなわちCAD(ほとんどのケース)、MDA、またはRRD。7年後、1/3しかremissionとは考えられなかった。65%は再発性か持続性の疾患活動性を有した。
9.1. Early treatment and the window of opportunity in SLE
SLEの管理における臨床的な推奨は十分な治療の開始の必要性である事である点においては全会一致である事は注目に値する。臓器に関わらず、どの薬剤が治療に提案されていようと。前述のように治療が早い程、持続鉄器寛解の達成のチャンスが高くなり、患者の予後を改善させる。人間における早期の介入には制限があるが、既存の研究は治療の遅れはよりヘビーな治療の必要性につながり患者のより悪いoutcomeに関連する。とくに腎炎において3-5ヶ月の診断・治療の遅れは寛解を低下させ、ESRDの危険因子である腎炎再発の頻度の増加に関連した。LNにおいて早期の治療開始は長年の重大な問題である。治療開始前の6ヶ月より長い期間の症状は最も強いESRDの危険因子であることが分かっている。そのため完成した疾患や合併症(つまりwindow of oppotunity)を避けるために治療を開始しなければならな時間は少なくとも腎炎においては約3-5ヶ月と思われる。
9.2. Early diagnosis helps early treatment
早期診断は早期治療につながる。現在のところSLEの発症から診断までのタイムラグは約9ヶ月であり、1980年より以前の59ヶ月と比べると劇的に短くなっており、これはANAの測定が臨床プラクティスに導入されてから達成された。自己抗体は疾患が明らかになる数年前から出現しているようであり、そのため抗核抗体を高感度に検出できるようになったことで、診断に要する時間が短縮された。しかし現在の9ヶ月はwindow of opportunityのおおよそ2倍であり、臓器障害を悪化させる時間と言える。早期診断が臓器機能不全の回復を最大化させる。この目標のために最も妥当なツールは分類基準の適切な使用、SLEのbiomarkerの測定、そしてSLEの所見に気づく開業医の教育(彼らの患者の中から)である。
9.2.1. New SLE classification criteria do not help early diagnosis in SLE(略)
9.2.2. Lupus biomarkers (略)
9.2.3. The importance of primary care physicians(略)
重篤な感染症(2002例中75例[3.75%] vs 2001例中82例[4.10%]、差-0.35%、95%CI -1.55~0.85)、日和見感染症およびその他の注目すべき感染症(36例[1.80%] vs 50例[2.50%], -0.70%, -1.60 to 0.20)の発生はベリムマブとプラセボ群に共通してみられた。
メラノーマ以外の皮膚がん(4 [0.20%] vs 3 [0.15%]; 0.05%, -0.21 to 0.31)、その他の悪性腫瘍(5 [0-25%] vs 5 [0-25%]; 0.00%, -0.31 to 0.31)。
Except for a limited number of safety oversight personnel and the site pharmacist or designee who dispensed the blinded study drugs, all other study site personnel, patients, the sponsor, and the contract research organisation were masked to the treatment assignment. Treatment allocation could be unmasked by the investigator or treating physician in the event of a serious medical emergency or medical condition.
注釈:適格性を審査した患者数およびこの段階で除外された患者数は収集していないため、不明。AESI=adverse event of special interest(特に関心のある有害事象)。ITT=intention-to-treat。SAE=重篤な有害事象。*4019人の患者が無作為割り付けされたが、無作為化の詳細がデータベースに入力されなかったため、1人の患者は表示されない;その患者は試験薬を受け取らなかった。1名の患者はプラセボ群に割り付けられたが、誤って50%以上の期間でベリムマブを投与されたため、ITT解析ではプラセボ群、as-treated解析ではベリムマブ群(ここでは無作為化)として異なるグループに含まれた。‡site closureによる。§ITT集団には、無作為に割り付けられた治療群に従って、少なくとも1回分の試験薬を投与された患者を含み、実際に投与された薬に関係なく、ITT集団に含まれた。患者が最も多く(50%以上)受けた実際の治療により、as-treated集団に含まれる。
A target population of 5000 patients was originally planned to give ±0·46% precision of the 95% CI for the between-group treatment difference for mortality, assuming a first-year mortality rate of 0·68% in both treatment groups. After the study began and following the completion of two additional randomised controlled trials of belimumab in SLE,3,10 the mortality rate estimate was adjusted to 0·56%, leading to a revised target population of 4000 patients to provide ±0·46% precision for the between-group treatment difference 95% CI for mortality. The request to change the sample size was made in December, 2016, when 304 sites from 33 countries had recruited 3459 participants. The randomised population was defined as all patients who were randomly assigned, with patients grouped according to the treatment that they were allocated to receive, regardless of actual treatment received. Safety analyses were done for the as-treated population (grouped according to the treatment that was received for more than 50% of doses), as the primary endpoints were safety endpoints. 当初、両群の1年目の死亡率を0.68%と仮定し、死亡率の群間差の95%信頼区間の精度を±0.46%とするために、5000名の患者さんを対象として計画されました。しかし、本試験開始後、SLEにおけるベリムマブの2つの無作為化比較試験3,10が終了したため、死亡率の推定値が0.56%に修正され、死亡率の群間差95%信頼区間の精度が±0.46%となるよう、対象患者数が4000名に修正された。サンプルサイズの変更要請は、33カ国304施設が3459人の参加者を募集した2016年12月に行われた。無作為化集団は、無作為に割り付けられたすべての患者と定義され、患者は実際に受けた治療にかかわらず、割り付けられた治療に従ってグループ分けされた。安全性解析は、主要評価項目が安全性エンドポイントであることから、As-treated集団(50%以上の投与で受けた治療法に従ってグループ化)を対象に行われた。
★結果の評価
全ての死亡はbelimumab 0.5% vs placebo 0.4% で、その差0.1%: 95%CI, -0.31 to 0.51%と差はなし